高质量的植物基因组序列对研究植物进化、育种和保护至关重要。然而,植物基因组可能非常大,而且高度重复,因此在传统技术下很难进行基因组组装。
Pore-C 是一个结合染色质构象捕获 (3C) 和纳米孔长读长测序的端到端工作流程,可提供接点信息,从而将现有组装搭建为高度连续的 scaffold。传统 3C 方法通常仅检测两个基因座之间的成对相互作用,而 Pore-C 可从多个基因座中生成高阶接点信息。该方法无需扩增,能够进入 GC 富集和重复基因组区域。表观遗传修饰也保留在纳米孔长读长序列中,可以与核苷酸序列一起得到表征。将该方法与高产出 PromethION™ 测序芯片共同使用,就可以实现高度连续的染色体规模植物基因组组装。
我们在此展示了一个完整的工作流程,用于搭建植物基因组组装的 scaffold,使其达到染色体规模。
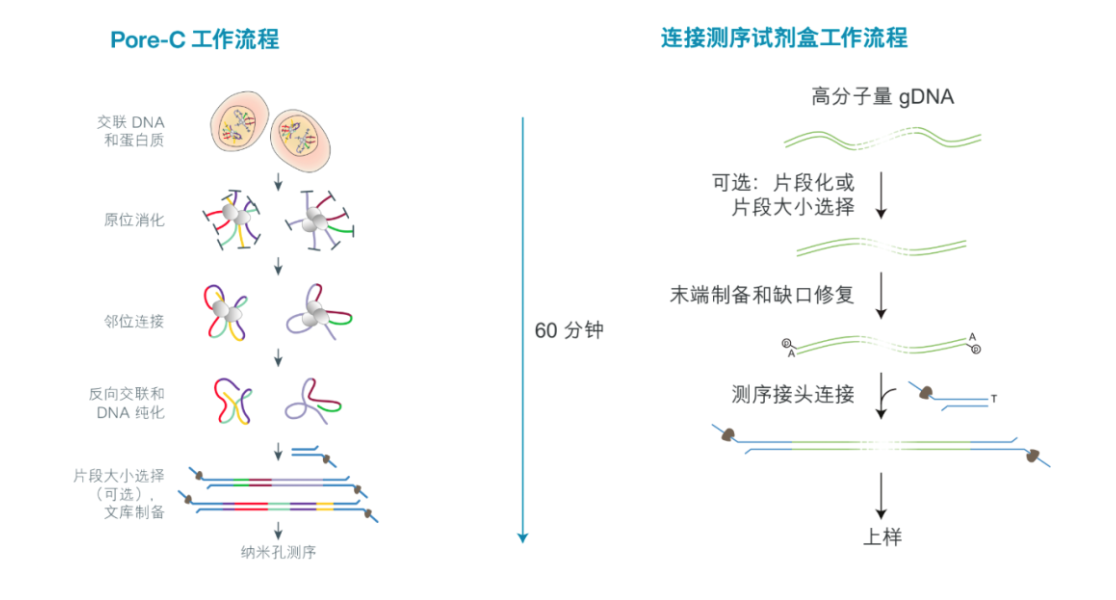
PORE-C 工作流程:
交联、DNA 提取和文库制备
从一片植物叶子样本开始,植物 Pore-C 工作流程开展了交联、消化和邻位连接工作:一种对 3D 空间中(但不一定在线性基因组序列中)紧密连接的基因座进行修复的流程。反向交联后,纯化样本。为了使包含接点的测序读长序列数量最大化,我们建议在文库制备之前使用 SPRI 磁珠,以选择大小超过 1.5-2 kb 的已连接片段。然后使用连接测序试剂盒制备 Pore-C 文库。但是,如果提取的 DNA 产量 <1 μg,我们建议使用快速聚合酶链反应 (PCR) 条形码试剂盒,以获得最高测序通量。
了解更多关于 Oxford Nanopore 测序试剂盒的信息:store.nanoporetech.com/sample-prep.html
有关 Pore-C 工作流程的详细指导,包括交联、DNA 提取和文库制备,请访问:
community.nanoporetech.com/info_sheets/restriction-enzyme-pore-c
测序:使用 PromethION 生成高产量的长读长序列
为了将现有植物基因组组装搭建为染色体规模的scaffold,我们建议将 PromethION 测序芯片上的Pore-C 文库测序至最低覆盖深度 20x。对许多植物基因组而言,在一张 PromethION 测序芯片上对一个Pore-C 文库测序 72 小时,即可实现这一目标。
使用测序芯片清洗试剂盒清洗测序芯片,然后大约每18 小时加载新的文库,可使通量最大化。
了解更多关于 PromethION 测序设备的信息:
nanoporetech.com/products/promethion
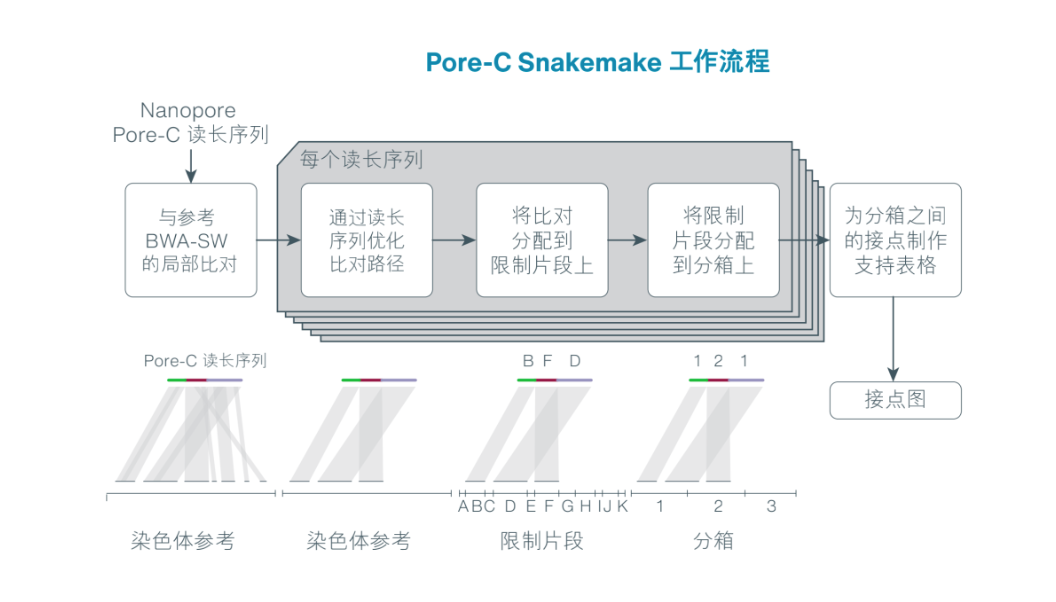
建议使用我们全面的 Pore-C Snakemake 工作流程(可在 GitHub 上获取)进行分析。
使用该工作流程时需要熟悉命令行。用 Pore-C 读长序列创建接点图,然后将读长序列与参考基因组进行比对。
然后,建议使用工具 Salsa21,利用上述信息找到重叠群位置,这样可以搭建 scaffold 并纠正之前的错误组装。使用这种方法,就可生成高度连续的染色体规模植物基因组组装。
查看 GitHub 上的 Pore-C 分析流程:
github.com/nanoporetech/pore-c
如需了解更多信息,请访问:
nanoporetech.com/applications/investigation/chromatin-conformation
1.Ghurye, J. et al. PLoS Comput Biol. 15(8):e1007273 (2019).
Oxford Nanopore Technologies、风轮图标、EPI2ME、Flongle、GridION、Metrichor、MinION、MinKNOW、PromethION、SmidgION、Ubik 和 VolTRAX 是 Oxford Nanopore Technologies plc 在不同国家的注册商标。包含的所有其他品牌和名称均为其各自所有者的财产。© 2022 Oxford Nanopore Technologies plc。版权所有。Oxford Nanopore Technologies 产品并非旨在用于健康评估或诊断、治疗、缓解、治愈或预防任何疾病或状况。
WF_1149(CN)V1_14Mar2022
 027-62435310 |
027-62435310 |
 service@speedracings.com |
service@speedracings.com |