 027-62435310 |
027-62435310 |
 service@speedracings.com |
service@speedracings.com |
Genome Research丨周扬与George E. Liu团队合作完成目前世界最大规模牛泛基因组和基因组结构变异数据库构建
近日,华中农业大学杨利国教授团队的周扬副教授与美国农业部George E. Liu团队合作在Genome Research杂志发表题为“Assembly of a pangenome for global cattle reveals missing sequences and novel structural variations, providing new insights into their diversity and evolutionary history”的论文,并被选为杂志的封面文章。该文章鉴定出牛参考基因组之外的83Mb新序列,率先从群体水平构建了牛泛基因组;同时开发基因组结构变异(Structural Variation, SV)分析流程,首次从SV的角度解析了世界牛品种的血统关系,从适应性等多角度全方位检测了不同分类群体的受选择位点和候选基因,填补了SV作为牛育种材料的不足,并解析SV潜在形成机理促进对牛基因组演化的深入理解。

图1 Genome Research杂志2022年8月份期刊封面
参考基因组序列是保障一切与基因相关研究顺利实施的必要基础。2003年长达13年之久的人类参考基因组计划(85%完整度)为人类解析生命活动开启了新篇章,被誉为与阿波罗登月计划和曼哈顿计划同等重要的大事件。牛参考基因组于2009年发布,在之后的13年内不断对其进行填补和完善,并相继公布了近10个牛品种的参考基因组。但每个参考基因组几乎只包含单个个体基因组序列,忽略了个体之间序列的差异性,导致在科学研究过程中相当一部分基因组序列及其功能处于未知状态;同时已知基因组序列在不同个体之间存在SV,导致不同个体间基因组序列构成非常复杂,精准检测和分型一直是阻止其后续解析和应用的障碍。
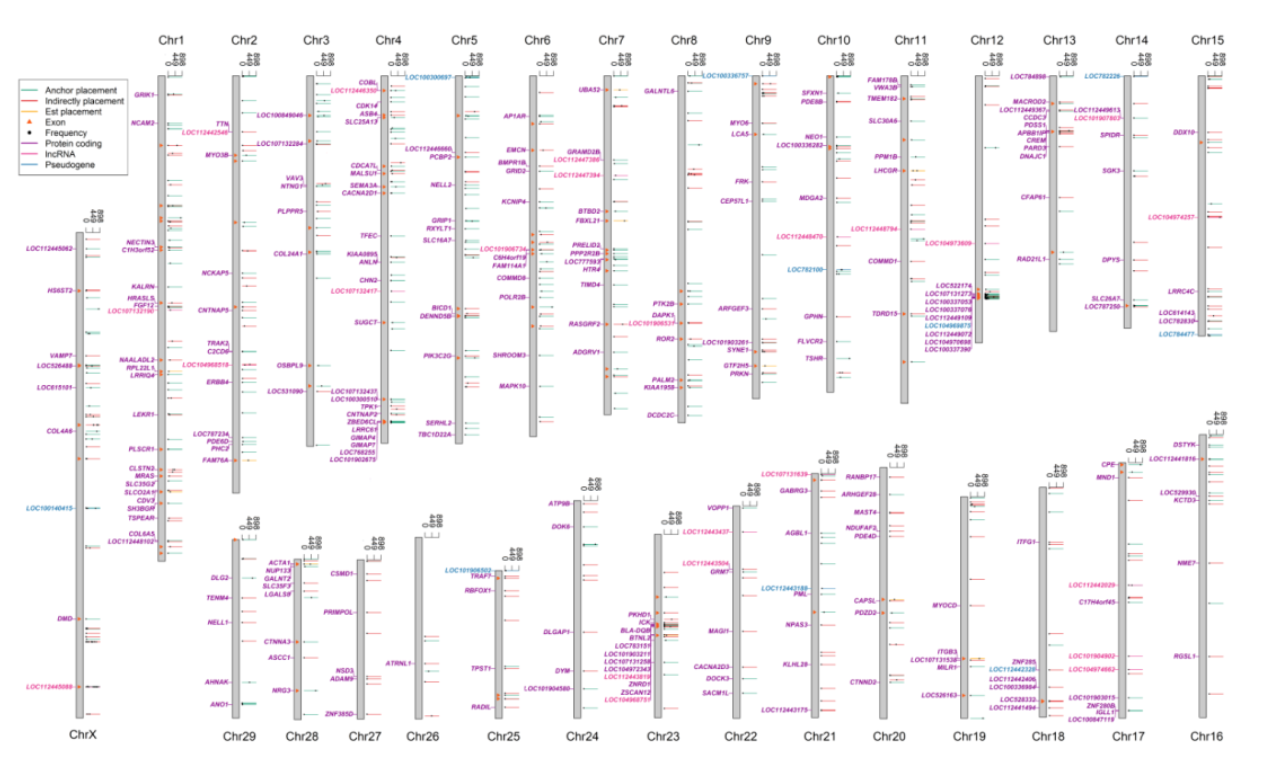
图2泛基因组新序列在已知参考基因组上的分布及影响
为解决以上问题,该文章通过开发新的基因组分析流程,对来自世界57个品种898头牛的基因组序列进行组装和SV检测;发现了83Mb的牛基因组未知序列,相当于目前参考基因组序列总长度的3.1%,并且从中鉴定出大量未知外显子序列,最后综合三种策略成功将1007条contig放置在现有参考基因组上,这些序列可能通过外显子破坏、外显子添加等方式影响了279个基因的序列结构。
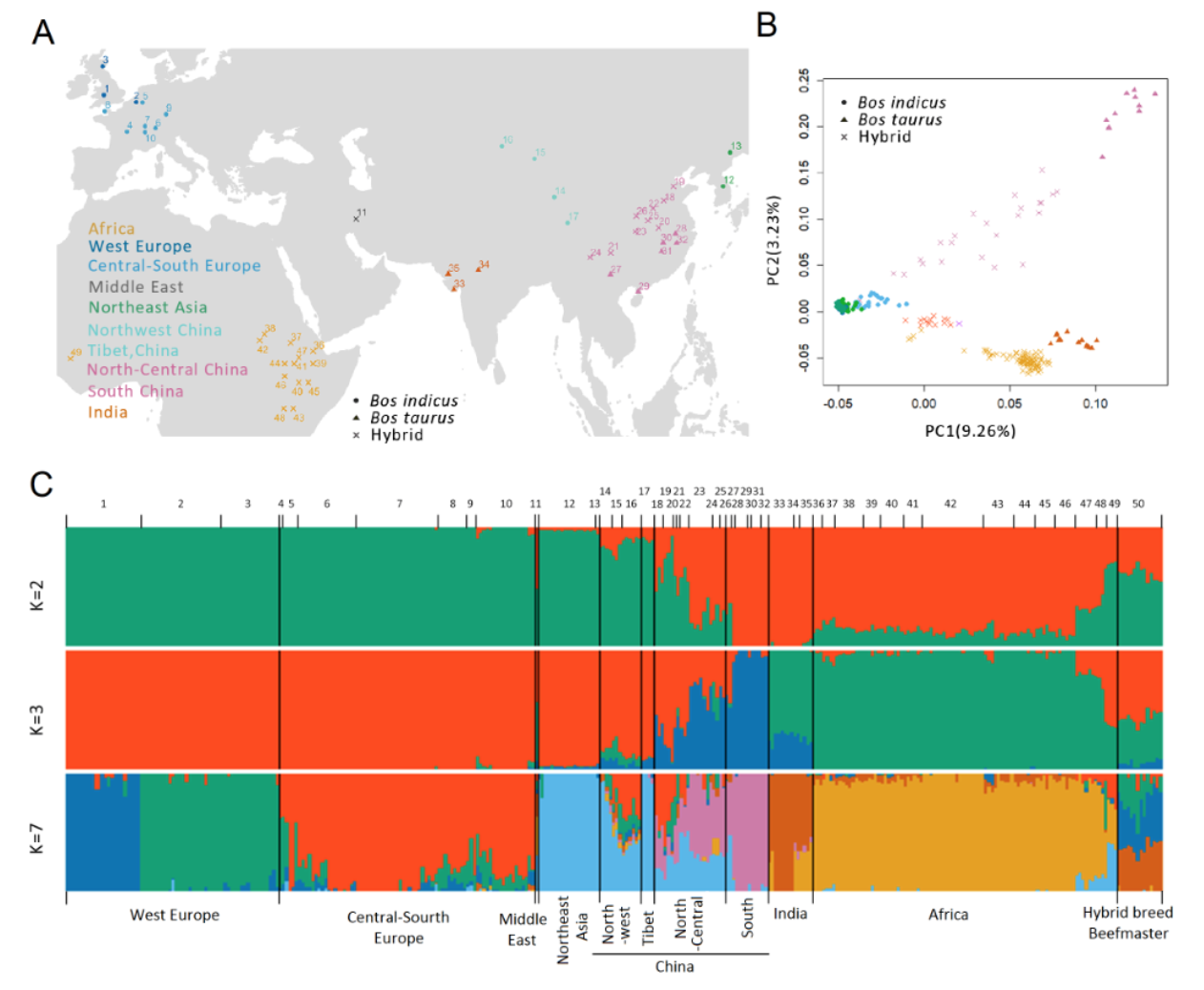
图3利用SV(缺失型变异)对世界牛品种展开精准群体学分析
同时,利用多款SV检测软件重新开发新的分析流程,在准确检测SV的基础上实现了对缺失型变异的边界精准鉴定和二倍体分型;利用此流程,共检测到了330万个缺失变异、12万个倒置、18万个重复区域,通过对检测结果的模拟评估,证明本论文所检测的SV在牛上已经趋于饱和,具有很好的群体代表性。以此为基础,完成了对SV对牛基因组27种不同基因组功能元件和QTL的影响研究,构建了受SV影响的基因功能元件数据库。进一步利用缺失型变异对世界牛品种的血统来源进行分析,实现基于SV水平的世界牛品种血统的精准鉴定,为解析世界牛品种血统组成提供了新方法,打破了长期使用SNP进行遗传学分析的局限。
进一步利用比较基因组学等手段对牛的种属差异、地域适应性、商业化用途等角度全面解析获得大量与经济性状相关的变异位点,并深入解析了位于APPL2第一外显子区域缺失变异与不同地域牛选择适应性之间的关系;发现该缺失变异是由Bov-tA1转座子构成,并形成了APPL2基因的启动子和增强子部分,该序列携带了APPL2相关功能的转录因子结合位点,通过影响基因的表达,进而调控与免疫反应、味觉功能、细胞增殖、葡萄糖代谢相关的功能。
周扬副教授和其硕士研究生杨旅同学为该论文共同第一作者,George E. Liu、杨利国教授和周扬副教授为共同通讯作者。该研究受到国家自然科学基金、湖北省自然科学基金、美国AFRI和NIFA等项目的资助。
英文摘要
A cattle pangenome representation was created based on the genome sequences of 898 cattle representing 57 breeds. The pangenome identified 83 Mb of sequence not found in the cattle reference genome, representing 3.1% novel sequence compared with the 2.71-Gb reference. A catalog of structural variants developed from this cattle population identified 3.3 million deletions, 0.12 million inversions, and 0.18 million duplications. Estimates of breed ancestry and hybridization between cattle breeds using insertion/deletions as markers were similar to those produced by single nucleotide polymorphism–based analysis. Hundreds of deletions were observed to have stratification based on subspecies and breed. For example, an insertion of a Bov-tA1 repeat element was identified in the first intron of the APPL2 gene and correlated with cattle breed geographic distribution. This insertion falls within a segment overlapping predicted enhancer and promoter regions of the gene, and could affect important traits such as immune response, olfactory functions, cell proliferation, and glucose metabolism in muscle. The results indicate that pangenomes are a valuable resource for studying diversity and evolutionary history, and help to delineate how domestication, trait-based breeding, and adaptive introgression have shaped the cattle genome.


