 027-62435310 |
027-62435310 |
 service@speedracings.com |
service@speedracings.com |
项目文章|“常香粳1813“染色体水平基因组揭示香粳和非香粳稻间的基因组变异!
研究简介
8月26日,江苏省中科院植物研究所孙小芹研究员联合常熟市农业科学院研究所、广东省林业科学院等单位研究团队,在期刊INTERNATIONAL JOURNAL OF MOLECULAR SCIENCES(IF=6.208)在线发表“Chromosome-Level Genome Assembly of a Fragrant Japonica Rice Cultivar ‘Changxianggeng 1813’ Provides Insights into Genomic Variations between Fragrant and Non-Fragrant Japonica Rice“研究论文,组装出“常香粳1813“的高质量染色体水平基因组并开展了比较基因组学等其他一系列分析工作。AG代理基因承担了该研究中的测序组装工作及部分分析工作。东亚拥有丰富的香粳稻资源,消费者和生产商对此都越来越感兴趣。然而,目前用于香粳稻育种的基因组资源,特别是全基因组序列仍然稀缺。本研究通过整合Nanopore长读测序、Illumina短读测序和Hi-C数据,组装得到了“常香粳1813“的高质量染色体水平基因组。基于注释数据,证明了badh2-E2型缺失是”常香粳1813“的香味来源。此外,作者还进行了比较基因组分析,进一步支持了”“BADH2在水稻的非生物胁迫耐受性中不起作用 “这一发现。最后,研究者选择了与”常香粳1813“基因组具有高度同源性的非香粳稻日本晴进行了基因组变异研究,结果,鉴定出了大量的SNP、INDEL以及SV。总之,这些基因组资源将有助于阐明重要经济性状的机制,并对芳香粳稻基因组学辅助育种具有广泛意义。

文章题目:Chromosome-Level Genome Assembly of a Fragrant Japonica Rice Cultivar ‘Changxianggeng 1813’ Provides Insights into Genomic Variations between Fragrant and Non-Fragrant Japonica Rice
发表期刊:INTERNATIONAL JOURNAL OF MOLECULAR SCIENCES(IF=6.208)
发表时间:2022.08.26
主要研究结果
1.基因组测序和从头组装
本研究中,作者通过结合Nanopore、Illumina和Hi-C技术的混合策略,对“常香粳1813”的基因组进行了测序和从头组装。过滤后,分别产生约51.59 Gb Nanopore长reads、约28.21Gb Illumina短reads和约41.45Gb Hi-C reads。基于Nanopore长reads进行初步组装,随后使用Nanopore和二代数据进行两轮纠错后,组装得到大小为378.78Mb、Scaffold N50高达29.83Mb的基因组。最后使用Hi-C数据辅助组装该基因组,最终将其挂载到12条染色体上,挂载率100%,成功组装出”常香粳1813“的染色体水平基因组。此外,二代reads与该参考基因组的比对率高达99.11%,完整的BUSCO为96.16%,表明本研究得到的”常香粳1813“组装表现良好且高度完整(图1)。
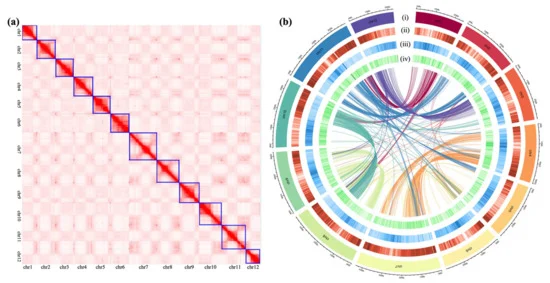
图1 “常香粳1813“基因组的基本特征
2.基因组注释
以往的研究表明,重复序列在基因组大小变异和功能适应中的作用,使得其在植物基因组进化中发挥着关键作用。该研究对基因组进行注释,发现50.52%的“常香粳1813”基因组被鉴定为转座元件(TEs),其中,DNA转座子最丰富,占基因组的24.47%,其次是长末端重复(LTR;24.15%),此外还有0.97%被鉴定为串联重复。在这里,作者还将疣粒野稻与“常香粳1813”的基因组大小和LTR比例进行了对比,并推测二者间LTR比例的差异与基因组大小相关。此外,该研究还预测到了32165个蛋白质编码基因、3524个非编码RNA,其中包括miRNA、tRNA、rRNA、snRNA等。
3.BADH2基因的特征及进化分析
由于“常香粳1813”已被鉴定为香米((2AP content:~310 ug/kg),故该研究还检查了它是否携带隐性BADH2基因,并研究了其等位基因变异。与非香粳稻品种日本晴相比,在BADH2等位基因(BADH2-E2)的第二外显子中观察到7bp的缺失,从而促进了2AP的积累。此外,该研究提到,badh2-E2等位基因与许多中国香粳稻品种中的等位基因一致,而且,BADH2单倍型数据的系统发育分析显示,“常香粳1813”中的BADH2-E2等位基因与先前确定的两种栽培粳稻特有的单倍型序列聚集在一起。这表明,该等位基因在中国香粳品种中普遍存在,但若想全面检测香米中badh2-E2的起源和演变,仍需更多样本的分析工作。
4.基因家族进化与系统发育关系
基因家族的扩展和收缩通常被认为是重要的进化机制。为了揭示“常香粳1813”中与环境胁迫相关的基因家族的扩张和收缩,该研究对稻属不同成员的基因家族规模进行了计算分析。结果表明,“常香粳1813”基因组中的896个基因家族经历了扩张,而1467个基因家族经历了收缩。功能富集分析发现,扩张的基因家族主要富集在与氧化应激等其他过程相关的成员上。由于氧化应激被视为暴露于各种非生物应激植物中的主要损害因素,因此,这些扩张的氧化应激反应基因可能在快速气候变化期间增强“常香粳1813”的应激耐受性。这一结果也支持了先前的发现:即BADH2在水稻的非生物胁迫耐受性中不起作用,并且没有与香味表型相关的非生物应激耐受性的普遍丧失。
5.“常香粳1813“与日本晴的基因组变异
与日本晴基因组相比,该研究在“常香粳1813“基因组中鉴定出289970个SNP和96093个INDEL,另外,SNP和INDEL在染色体间和同一染色体内的分布不均匀,且二者的分布呈正相关,在基因间间隔区更为丰富,更具体地说,多数SNP和INDEL分布在内含子中(图2)。
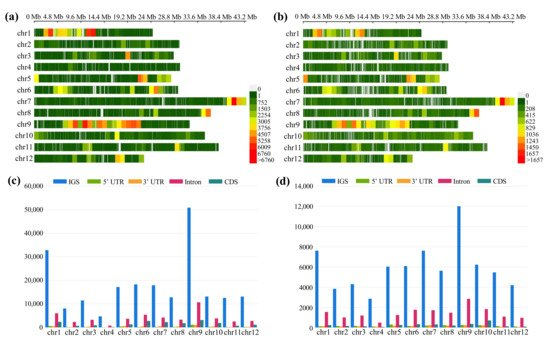
图2 Distribution patterns of SNPs (a) and InDels (b) across the ‘Changxianggeng 1813’ genome by comparing to the Nipponbare genome. (c,d) The distribution of (c) SNPs and (d) InDels in different genomic regions, including intergenic spacer regions (IGS), 50 untranslated regions (UTR), 30 UTR, intron and protein coding regions (CDS).
另外,该研究基于Nanopore长reads的优势,在”常香粳1813“和日本晴基因组间鉴定到了8690个SV,其中DUP是占比最高的一类。最后,作者还研究了SVs在不同基因组区域的分布,发现70%的SV都位于非编码区。以往的研究表明SVs重叠基因可能影响基因的功能和表达,本研究发现SVs重叠基因在每条染色体上的分布不均匀,证明了某些区域可能是保守的,并且在”常香粳1813“和日本晴之间共享一个共同的祖先基因库(图3)。
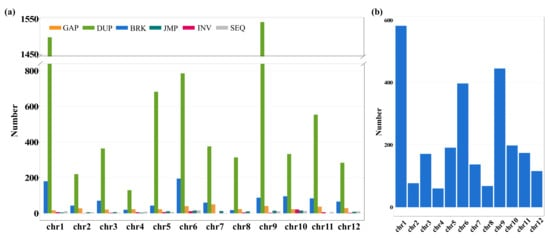
图3 (a) SV types and numbers across 12 chromosomes of the ‘Changxianggeng 1813’ genome. (b) Total counts of SVs overlapping genes for each chromosome in the ‘Changxianggeng 1813’ genome. GAP , gap between two mutually consistent alignments; DUP , inserted duplication; BRK, other inserted sequence; JMP , rearrangement; INV , rearrange ment with inversion; SEQ, rearrangement with another sequence.
研究结论
综上,研究者使用Nanopore、Illumina和Hi-C测序数据的组合,提供了一个新的香稻品种“常香粳1813”的高质量参考基因组序列,这是芳香粳稻的第一次从头染色体水平基因组组装。此外,还基于该注释的基因组序列,证明了badh2-E2型缺失导致了该粳稻品种的香味。最后,还对“常香粳1813”和非香粳稻日本晴的基因组进行比较以鉴定基因组变异。毫无疑问,这些基因组资源将促进水稻的基因和基因组研究,并有利于香粳稻的品种改良。
原文链接://www.mdpi.com/1422-0067/23/17/9705


