 027-62435310 |
027-62435310 |
 service@speedracings.com |
service@speedracings.com |
文献解读| Nature microbiology封面文章—内蒙古人肠道微生物群的高质量基因组简编
基于宏基因组的资源已经揭示了人类肠道微生物群的多样性和功能,但由于基因组质量不足,缺乏典型的研究人群,限制了进一步的理解。本研究使用混合长读PromethION和短读HiSeq测序来表征内蒙古人的粪便微生物群。研究最终提出了内蒙古人肠道基因组目录,包括802个闭合的和5927个高质量的宏基因组组装基因组。超深度混合测序策略实现了高度的基因组连续性,大幅提高了基因组元件的分辨率。尤其是报告了未培养物种的核糖体RNA操纵子拷贝数,超过12000个以前未描述的肠道前噬菌体和插入序列元件。总之,本研究扩展了目前的人类肠道基因组目录,内蒙古肠道基因组数据集(IMGG)为研究人类肠道菌群提供了高质量、大规模的资源。

文章标题:A high-quality genome compendium of the human gut microbiome of Inner Mongolians
发表期刊:Nature Microbiology (IF=30.964)
发表时间:2023.01.05
研究策略
1. 干预试验设计和参与者招募
招募了160名居住在呼和浩特的卫生志愿者,在第0、7和28天采集粪便样本,随机选择60名参与者(30名饮用普通酸奶,另外30名饮用益生菌酸奶)的粪便样本(共180份)用于后续宏基因组测序
2. Nanopore和Illumina文库制备及测序
在Oxford Nanopore PromethION和Illumina HiSeq 2500测序平台上进行宏基因组测序,共生成3.7 Tb长读和20.1 Tb短读数据,每个样本的长读数据为20.5±4.5Gb,短读数据为111.8±8.65 Gb
3. 生物信息学分析
l Nanopore序列处理,宏基因组组装和分箱
l UHGG和IMGG数据集中基因组的种级聚类分析
l 基因组质量与比较基因组学分析
l 前噬菌体挖掘、聚类,分类和功能注释
l IS元件及IS元件连锁基因的挖掘与分析
主要研究结果
1. 超深混合宏基因组测序和组装
本研究的长读数据集的质量远远超过现有的大多数人类肠道长读数据集,平均N50长度为8 Kb。混合超深宏基因组测序获得了平均N50长度为278 Kb的宏基因组组装。最大contig为6.77 Mbps, 6688个contig大于1 Mbps。同时使用三种测序和组装策略直接从摄入益生菌酸奶的个体的粪便样本中组装Probio-M8基因组:长读修正、长读加短读修正以及短读组装。不出意料的是,与用长读或短读测序单独构建的基因组相比,长读加短读修正基因组在多个基因组质量参数上都有很大的改善,包括组装邻近性、碱基对和基因预测精度、基因组完整性等。表明长读和短读纠错是直接从复杂的人类肠道宏基因组数据集中组装闭合和精确基因组的有效策略。
2. 高效组装大量的种级别CMAGs
混合宏基因组数据的高质量组装共产生了802个CMAGs(完整的MAGs)。为了评估目前组装的CMAGs的代表性,将物种水平去重复后的数据集与人类胃肠道基因组(UHGG)集进行了比较。总共保留了134个种级CMAGs,涵盖11门、14纲、27目、45科、94属(图1)。特别是,131个种级CMAGs是该物种(截至2021年7月)未报道的环状、完整、人类特异性的代表性基因组,65个未培养物种的参考基因组质量得到提高。
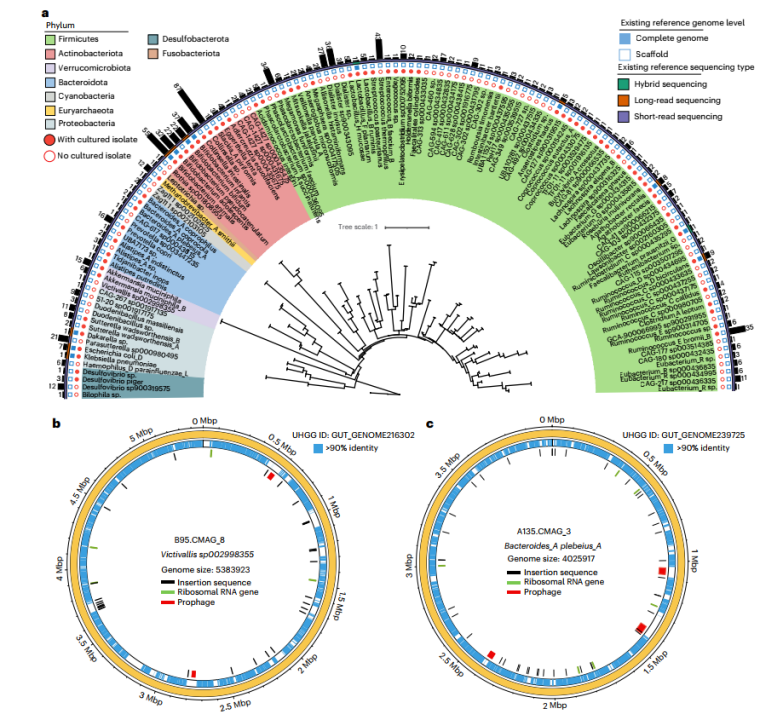
图1 高效组装大量的种级别CMAGs
3. 内蒙古人高质量肠道基因组目录
对IMGG数据集中的6729个高质量MAGs和UHGG数据集中的147835个高质量基因组进行聚类分析(图2a)。经分类鉴定,共归属11门14纲30目40科220属(图2b),72.7%属于厚壁菌门。在485个MGS中,有469个是同时存在于UHGG和IMGG数据集的人类肠道中高丰度的物种。其余16个MGS在IMGG数据集中是唯一的,一半属于梭状芽孢杆菌目。
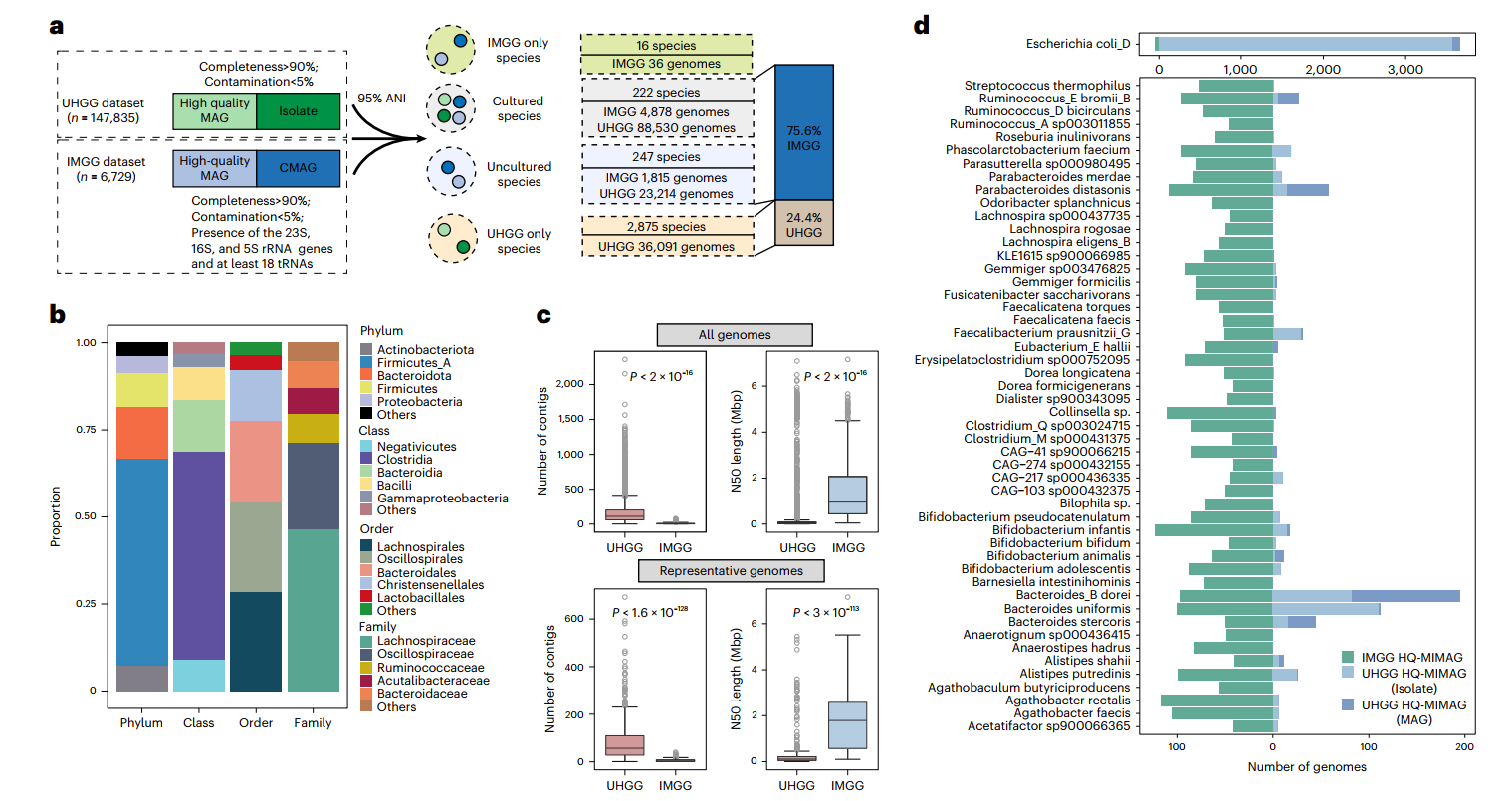
图2 IMGG目录是一个扩展的基因组资源
4. 提高了复杂肠道基因组区域的分辨率
为了揭示复杂的肠道基因组区域(包括rrns、MGCs、前噬菌体和IS元件),从IMGG数据集组装的485个MGS与UHGG数据集的对应物种进行了比较。IMGGs有更多的rrn拷贝(图3a),IMGGs与NCBI中对应的完整分离基因组的rrn拷贝数基本一致。其次,比较IMGGs和UHGGs中代谢基因簇(MGCs)的完整性。结果显示,在UHGGs中鉴定出的大部分MGCs位于contig边缘,可能是不完整的,而在IMGGs中仅为4.5% (图3b)。IMGGs中包含MGCs编码序列的基因组比例显著高于UHGGs (图3c)。另外,与UHGG数据集相比,IMGG数据集的前噬菌体基因组分辨率显著提高,在95%的IMGGs中检测到前噬菌体序列(而在UHGGs中检测到55%)。与UHGGs相比,IMGGs的IS区域更加细化,每种IS区域的总拷贝数均显著增加(图3h)。
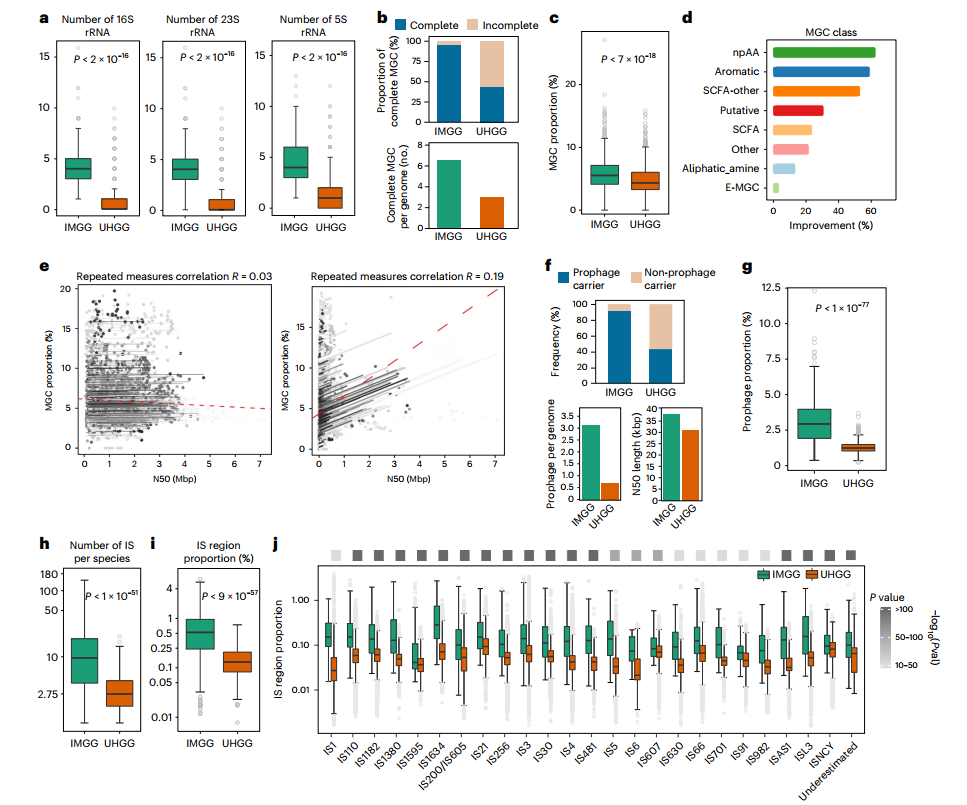
图3 增强IMGG基因组中遗传元件的分辨率
5. 人类肠道微生物中高度多样化和不同的MGCs
本分析只纳入UHGG和IMGG数据集中的高连续性和高质量基因组。从15512个基因组中共恢复了97428个MGCs区域,其中78675个是完整的。48%的完整MGCs与已知的MGCs没有同源性。门级分布和聚类分析显示,MGC类分布总体上有显著的门间差异(图4a)。富集/消耗分析一致地揭示了优势门中某些功能类的丰度存在显著差异(图4b)。Lachnospirales和Oscillospirales两个优势目包含最多的SCFA -生物合成途径(图4c)。这些数据表明,肠道微生物组MGCs编码了广泛的代谢潜能,不同关键的人类肠道分类群的代谢潜能差异很大。
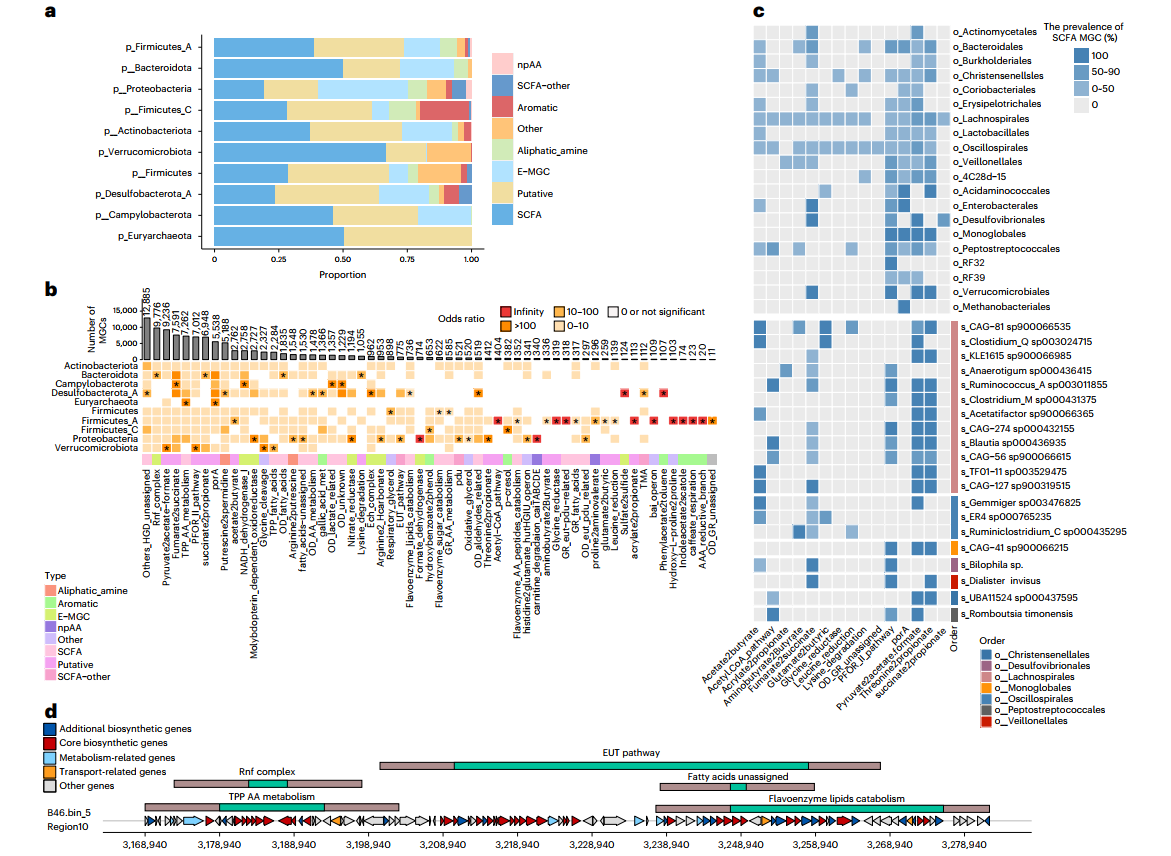
图4 人类肠道微生物群中的MGC池概述
6. 肠道前噬菌体的分类和功能注释以及IS元件的分布和作用
从IMGG数据集中恢复了21217个前噬菌体基因组,这些基因组进一步聚类为13437个种级病毒操作分类单位(vOTUs)。通过与宏基因组肠道病毒(MGV)目录比较,使用科级和种级系统发育距离来对vOTUs进行分类分配。95.5%的vOTUs与MGV目录没有种级同源性。90.9%的vOTUs与MGV目录具有科级同源性,但其中只有46.7%的vOTUs被分配到已知的病毒科(图5a)。以虹吸病毒科和肌病毒科为主(图5 b),并且具有广泛的宿主范围,跨越多个门(图5c)。从种级vOTUs基因组中鉴定出596193个蛋白质编码基因,55.4%的前噬菌体基因被功能分类,而44.6%被分配到未知的功能(图5d)。大比例的未匹配基因代表着病毒功能基因和代谢能力的巨大未开发库。
从公共数据库中检索的肠道细菌分离株的可用完整/染色体水平基因组和IMGG数据集中基于长读的MAGs中识别并编译了一个全面的IS元件列表。共鉴定出140336个IS元件(属于26个IS科)。不同的IS元件家族分布在多个门(图5i)。Proteobacteria和Campylobacterota在所有门中分别拥有最多和最少的IS元件拷贝数(图5i)。为探索转座元件的功能能力,对乘客基因(位于IS元件边界内)和邻近基因(位于IS元件附近)进行了分析和注释。结果表明,20.2%的IS元件编码至少一个假定的功能基因,乘客基因总数为36308个。UniProt数据库注释显示,这些乘客基因在DNA整合、细胞活动和转录调节等方面发挥着重要作用(图5k)。这些结果强调了一个事实,即肠道微生物相关的IS元素在分类和功能上都是多样化的。
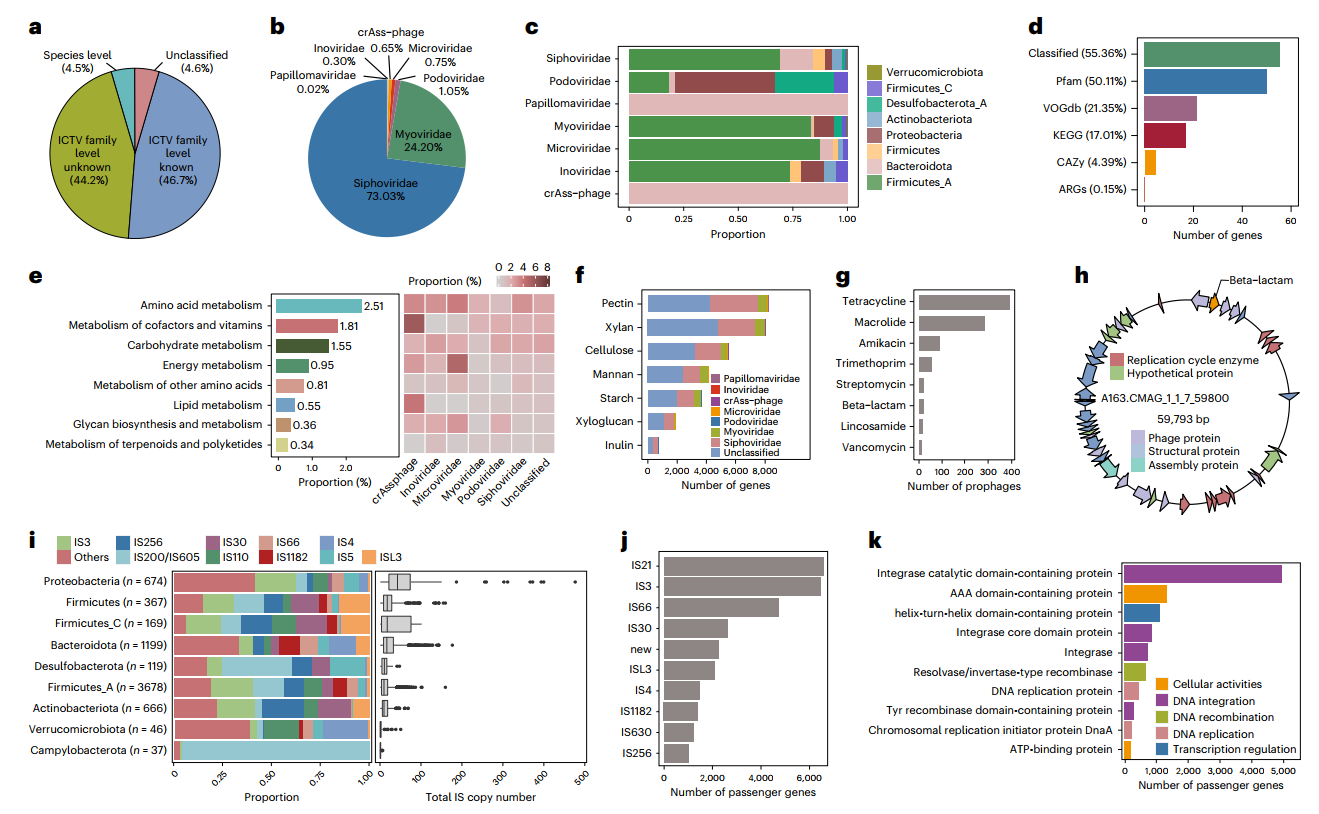
图5 简要介绍未描述的肠道噬菌体和IS元件
全文总结
总的来说,本研究结果支持超深短读polish是一种有用的策略,可以提高直接从复杂的人类肠道MAGs的连续性、准确性和分辨率。IMGG数据集是一个巨大的高质量基因组集合,具有未被充分代表的基因组区域的详细视图。作者设想,IMGG和其他大规模的高质量基因组目录在未来可以被整合,以创建统一质量的基因组存储库,进一步实现基因组解析宏基因组学。
参考文献:Jin, H., Quan, K., He, Q. et al. A high-quality genome compendium of the human gut microbiome of Inner Mongolians. Nat Microbiol 8, 150–161 (2023).


